Edema periorbitario recurrente
RESUMEN
Presentamos un paciente con episodios de angioedema palpebral de repetición, junto con aparición posterior de debilidad de la musculatura proximal; el diagnóstico final tras realizar el estudio complementario fue de miopatía inflamatoria tipo polimiositis.
Edema periorbitario recurrente
Caso clínico
Doctora Ana Belén Cid Sánchez, Médico de familia; Especialista en Alergología e Inmunología Clínica.
Palabras clave: edema, polimiositis
Se presenta el caso clínico de un varón de 40 años de edad con episodios de angioedema palpebral recidivante. Sin antecedentes personales ni familiares de interés.
Refiere desde hace un año episodios de edema palpebral recurrente que empeora con el decúbito. Progresivamente esta clínica se había ido agravando hasta afectar a cara, manos y pies; sin apenas mejoría con el tratamiento antihistamínico que había realizado.
A los cuatro meses del inicio de dicha clínica comenzó a presentar astenia y mialgias tras pequeños esfuerzos, más acusados en la musculatura de miembros inferiores junto con dificultad para la elevación de los miembros superiores por encima de la cabeza.
Exploración física:
Leve limitación de la movilidad de las articulaciones escápulo-humerales con fuerza conservada. Sin lesiones cutáneas
Pruebas complementarias:
- Hemograma: normal.
- VSG: 30 mm por segundo.
- Proteinograma: normal.
- Coagulación: normal.
- Perfil tiroideo: normal.
- Bioquímica: GOT 70 GPT 100 LDH 700.
- Serología negativa para citomegalovirus (CMV), virus de la hepatitis B (VHB), virus de la hepatitis C (VHC) y positivo para mononucleosis (IgM VCA, IgG VCA, IgG EBNA).
- Complemento C3, C4, C1 inhibidor, C1q y CH50 normal
- Factor reumatoide: negativo.
- PCR: normal.
- Enzimas musculares: CPK 5700(rango normal 5-195), aldolasa 180 U/l (rango normal 0-7,6).
- Inmunoglobulinas (IgA, IgG, IgE): normales.
- Crioglobulinas: negativas
- Anticuerpos antifosfolípido: negativos.
- Anticuerpos antimúsculo liso: negativos.
- Anticuerpos antimitocondria: negativos.
- Inmunocomplejos: negativos.
- ANCA: negativos.
- ENA: negativos
- ANA 1/2560 patrón nucleolar.
- Ecografía abdominal: normal
- Electromiograma: signos de miopatía difusa más expresiva en territorio proximal de ambas extremidades, con signos de lesión activa (denervación en reposo) de grado moderado.
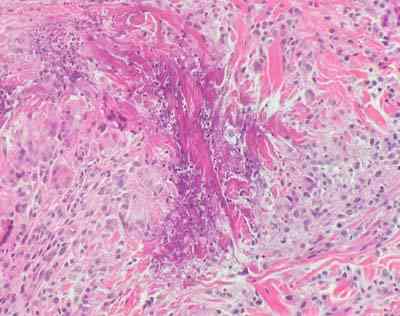
- Biopsia muscular: necrosis aleatoria de algunas fibras musculares con un infiltrado inflamatorio de células mononucleares.
- Tras el estudio realizado se diagnosticó de miopatía inflamatoria tipo polimiositis.Se instauró tratamiento con metilprednisolona 40 mg vía intravenosa cada 12 horas con la consiguiente disminución progresiva de las enzimas musculares y una normalización analítica así como desaparición de la clínica muscular.
- DISCUSION
- La polimiositis pertenece a un grupo de enfermedades (miopatías inflamatorias) que se caracterizan por presentar un infiltrado inflamatorio local o difuso del músculo esquelético, además de necrosis de las fibras musculares.La etiología es desconocida, con un posible origen inmunitario en el que se produce una destrucción de la fibra muscular del tipo celular citotóxico mediado por linfocitos TCD8; el infiltrado celular invade la fibra muscular.Suele predominar en el sexo femenino en una proporción de 2:1, con una edad media de presentación de 40 a 50 años de edad.La manifestación clínica más importante es la debilidad muscular, que progresa de forma lenta y progresiva, y de manera aguda en casos raros. Casi siempre simétrica y proximal sin afectación a los músculos inervados por pares craneales. Afecta fundamentalmente a cintura pelviana, cintura escapular y región cervical. Puede producir disfagia, voz nasal y regurgitación nasal de los líquidos, si afecta a la musculatura estriada y del tercio superior de la faringe. Con atrofias musculares en fases tardías. No es habitual la afectación de músculos faciales y extraoculares.También están presentes los síntomas sistémicos: fiebre, pérdida de peso, astenia, fenómeno de Raynaud. A nivel pulmonar: neumonía por aspiración, infecciones oportunistas, enfermedad pulmonar intersticial. El compromiso cardiológico es poco frecuente, hay casos de arritmia, insuficiencia cardiaca y pericarditis. Se describen artralgias y artritis.El diagnóstico clínico se confirma con la elevación de las enzimas musculares, las alteraciones electromiográficas y los hallazgos histopatológicos.Desde el punto de vista de laboratorio encontramos una elevación marcada de los enzimas musculares: (CPK; aldolasa; GOT; LDH). La CPK (creatin Kinasa) es un indicador muy sensible y específico de enfermedad muscular. Es una enzima citoplasmática que ante una destrucción de la fibra muscular o una alteración en la permeabilidad de la membrana rápidamente eleva sus niveles en sangre.El tratamiento con inmunosupresores es efectivo y, por lo general, bien tolerado en la mayoría de los pacientes junto con glucocorticoides, sin olvidar la terapia física o de rehabilitación, incluso en la fase aguda. Se han utilizado diferentes regímenes de tratamiento; sin embargo, la mayoría sugiere la administración de altas dosis de prednisolona (60 mg/día) al inicio y, posteriormente, disminuir la dosis en días alternos.
BIBLIOGRAFÍA
- Bohan A. y Peter J.B. Polymyositis and Dermatomyositis. N. Engl J Med 1975; 292:344-347, 403-407.
- Choy EHS, Isenberg DA. Treatment of dermatomyositis and polymyositis. Rheumatology 2002; 41:7-13.
- Plotz P.H., Dalakas M.C., Leff R.L., et al. Current concepts in the idiopatic inflammatory myopathies: Polymyositis, Dermatomyositis and related disorders. Ann Intern Med 1989; 111:143-157.
- Dalakas M.C. y Hohlfeld R. Polymiositis and dermatomyositis. The Lancet, 2003; 362:971-982.
- Christopher-Stine L. y Plotz P.H. Myositis: an update on pathogenesis. Curr Op Rheum 2004; 16:701-706.
- Choy EH, Hoogendijk JE, Lecky B, Winer JB. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. 2005; CD003643.