jueves, 18 de diciembre de 2008
Cual es el diagnóstico? Síndrome de Horner
Paciente de 62 años que consulta por dolor en región interes capular irradiado a región de hombro izquierdo de 3 meses de evolución tratada con medicación antiinflamatoria y corticosteroides sin respuesta.
capular irradiado a región de hombro izquierdo de 3 meses de evolución tratada con medicación antiinflamatoria y corticosteroides sin respuesta.
Al ingreso al hospital Pintos, la paciente presenta una anormalidad facial.


Se llevó a cabo una biopsia escisional de adenomegalia de fosa supraclavicular que mostró metástasis de carcinoma indiferenciado de pulmón. En este momento se está realizando inmunomarcación de tejido ganglionar.
Es decir que se trata de un cáncer de pulmón que debutó con dolor dorsal ysíndrome de Horner izquierdo

El syndrome de Horner resulta de una interrupción del tono simpático al ojo y se caracteriza por la clásica tríada de miosis, ptosis parcial, y ausencia de sudoración hemifacial (anhidrosis)Origen del términoFrançois Pourfour du Petit (1664-1741) describió primero esta condición en 1727 en animales de experimentación cortando los nervios en el cuello de perros y notando los trastornos que ocurrian en el ojo y la hemicara homolateral a la lesión neurológica. (1) En 1838 el clínico ingles Edgard Selleck Hare (1812-1838) hizo una descripción de un paciente que murió de un tumor en la región cervical. Fue mas profundamente descripto por Claude Bernard en 1852 (2)
Un reporte clínico del síndrome en un hombre que recibió un trauma en el cuello fue descripto en 1864 por los tres clínicos del ejército norteamericano: Silas Weir Mitchell (1829-1914), William Keen Jr. (1837-1932), and George Read Morehouse (1829-1905) (3). Mas tarde, en 1869, Horner describió una mujer de 40 años quien desarrolló las manifestaciones clásicas del síndrome (4) En 1958. D G Dirham documentó una familia en la que 5 personas en 2 generaciones fueron afectados (5) Esta rara condición genética es probablemente un rasgo dominante autonómico.
El término síndrome de Horner es comúnmente usado en paises ingleses, mientras que el término síndrome de Bernard-Horner es común en Francia.
El síndrome de von Passow es una asociación de síndrome de Horner con heterocromía del iris (6)
Patofisiología:La inervación simpática del ojo consiste en una vía de 3 neuronas. La primera neurona está en el hipotálamo ipsilateral y sus axones descienden por el tronco cerebral y la médula cervical hasta T1-T2. Esas fibras hacen sinapsis después de salir de la médula con el ganglio simpático cervical y hasta entonces son llamadas preganglionares. Una vez que hacen sinapsis con las células del ganglio cervical superior, las fibras de tercer orden viajan via carótida interna hasta la órbita, e inervan los músculos dilatadores del iris.
Las fibras simpáticas posganglionares también inervan el músculo de Mueller dentro del párpado. Este músculo es el responsable de iniciar la retracción del párpado durante la apertura palpebral. Las fibras simpáticas posganglionares, responsables de la sudoración facial siguen a la carótida interna hasta las glándulas sudoríparas de la cara. La interrupción en cualquier localización resulta en síndrome de Horner ipsilateral.
El síndrome de Horner puede ser el resultado de las siguientes condiciones:
* Lesión en la primera neurona (hipotálamo).
* Accidente cerebrovascular o tumor de tronco cerebral.
* Trauma de plexo braquial.
* Tumor (por ej Pancoast) o infección del vértice pulmonar.
* Lesión de fibras posganglionares.
* Disección carotídea (en un estudio 44% de los pacientes con disección de carótida extracraneal tuvo síndrome de Horner doloroso).
* Isquemia de la arteria carótida.
* Migraña.
* Neoplasia de fosa craneal media.
Frecuencia:El síndrome de Horner es poco común.
Morbi/mortalidad:Depende de la enfermedad de base.
Raza:No hay prerdilección por ninguna raza
Sexo: no hay predilección por sexo
Edad:No hay predilección por ninguna edad
Clínica:Los síntomas dependen de la enfermedad de base
Los pacientes pueden ser incapaces de abrir completamente el ojo afectado o de presentar trastornos de la sudoración en la hemicara afectada.
Los pacientes conlesiones preganglionares pueden tener enrojecimiento facial (flushing) (efecto arlequin) después de un ejercicio físico.
Los pacientes con lesiones posganglionares pueden tener dolor orbitario ipsilateral o cefalea de tipo “migraña like”
Raeder, un oftalmólogo noruego describió un paciente con combinación de dolor orbitario, miosis, y ptosis, síndrome que denominó paratrigeminal (9)
Cuando se asocia a disección carotídea los pacientes pueden presentarse con dolor ipsilateral en cuello, cabeza y cara
Los signos
Características pupilares:
La pupila del lado afectado puede ser redonda y contraida (miótica)
Los pacientes pueden tener pérdida del reflejo ciliospinal (aferente C2, C3). Las pupilas no se dilatan cuando la piel de la espalda es pinchada. Este hallazgo según muchos autores no es confiable
La anisocoria es mas evidente en la oscuridad. La pupila afectada se dilate mas lentamente que la pupila normal
Características faciales:
Los pacientes tienen la piel seca (anhidrosis) del mismo lado que la pupila afectada
Si la lesión es en la arteria carótida primitiva la pérdida de sudoración afecta toda la cara, y si la lesión es distal a la bifurcación la ausencia de sudoración está restringida a la frente y el lado de la nariz ipsilateral a la lesión
Otros hallazgos:
Los pacientes pueden tener ptosis parcial.
Pueden tener enoftalmos, aunque en general esto no ocurre, y la ptosis da la ilusión de enoftalmos, producido por la debilidad del músculo de Mueller, tanto en el párpado superior (causando ptosis parcial) como en el inferior
La amplitud de la acomodación aumenta.
La heterocromía del iris puede presentarse si la lesión es en un niño menor de 2 años. El iris afectado puede permanecer azul cuando el otro cambia a marrón. La pigmentación del iris está bajo control simpático durante el desarrollo, que es completado a los 2 años. La heterocromía del iris es infrecuente en pacientes con síndrome de Horner adquirido tardiamente en la vida.
Los pacientes pueden tener retracción del párpado contralateral .
Los pacientes pueden presentar disminución transitoria de la presión intraocular y cambios en la viscosidad de las lágrimas.
Los pacientes pueden no tener pliegue horizontal en el párpado ptosado especialmente en aquellos con síndrome de Horner congénito.
Los pacientes pueden tener enrojecimiento de la conjuntiva.
Causas:El síndrome de Horner puede ser congénito, adquirido, hereditario (autonómico dominante) La interrupción de las fibras simpáticas puede ocurrir en sistema nervioso central (entre el hipotálamo y el lugar de emergencia de la médula espinal (C8, T2), o en sistema nervioso periférico (por ejemplo en cadena simpática cervical, ganglio cervical superior, junto a la arteria carótida)
Causas centrales (raras)
El síndrome de Horner puede estar asociado con una lesión en el hipotálamo, hasta la médula en la región cervical superior. El síndrome medular lateral de Wallenberg (stroke), enfermedades desmielinizantes, y mas raramente trauma, o siringomielia pueden resultar en síndrome de Horner.
Es raro en el coma, pero el síndrome de Horner puede ocurrir ipsilateralmente a una gran hemorragia cerebral que afecta el tálamo. El síndrome de Horner ipsilateral en un paciente con parálisis laringea sugiere una lesión intramedular
El síndrome de Horner que ocurre en asociación con trauma medular sugiere lesión cervical alta, y no ocurre en lesiones por debajo de T2 o T3.
Causas periféricas:
El síndrome de Horner puede ser causado por lesiones en la cadena simpática, en el ganglio cervical superior, o acompañando a la arteria carótida como resultado de un tumor de Pancoast (carcinoma del vértice pulmonar), trauma, canulación de la vena yugular interna, o raramente de disección carotidea, trauma carotideo, colocación de un tubo en tórax para drenaje de derrame pleural (10), sarcoidosis, o tuberculosis en los ganglios cervicales (escrófula)
Un síndrome de Horner ipsilateral y neuropatía craneal afectando los pares craneales IX hasta XII puede estar asociado con compromiso carotideo, una rara pero fatal complicación de una infección del espacio faringeo.
La lesión común que causa síndrome de Horner, interfiere con las fibras preganglionares cuando ellas atraviesan la parte superior del tórax. Virtualmente todas las lesiones simpáticas posganglionares están localizadas intracranealmente o infraorbitariamente debido a que el ganglio cervical superior está cerca del cráneo.
El síndrome de Horner preganglionar indica una seria patología y con alta frecuencia se asocia a cáncer. Las causas comunes de síndrome de Horner incluyen trauma, disección aórtica, disección carotidea, y tumor de Pancoast, metástasis a los ganglios cervicales, y sarcoidosis o tuberculosis de los ganglios cervicales.
Un síndrome de Horner doloroso sugiere la posibilidad de disección de carótida interna.
El síndrome de Horner posganglionar tiene causas principalmente benignas (usualmente cefalea vascular). Las causas comunes de síndrome de Horner posganglionar incluyen trauma, cefalea histamínica y cirugía de cuello o de toroides
Diagnóstico diferencial
Anisocoria simple
Miosis senil
Pupila de Argyll Robertson.
Uso de drogas mióticas
Pupila de Adie-Holmes (contralateral)
Métodos de estudioImágenes:
Obtener una Rx de tórax para descartar un tumor del vértice pulmonar (carcinoma broncogénico apical) que es la causa más frecuente de síndrome de Horner.
Realizar TC de cráneo, si se sospecha ACV (stroke).
En el síndrome de Horner doloroso, obtener una angiografía por RMN con imágenes de sección cruzada del cuello para evaluar la posibilidad de disección carotidea
Procedimientos o maniobras diagnósticas
Los siguientes tests farmacológicos documentan la presencia o ausencia de una lesión simpática ocular e identifican el nivel de compromiso (preganglionar o posganglionar). Localizar la topografía de la lesión es importante debido a que las fibras preganglionares están asociadas a una mayor incidencia de cáncer que requiere investigaciones más profundas.
Tests que documentan la lesión ocular simpática
Test de la cocaína tópica:Agente: cocaína (2 a 4%)
Respuesta normal: dilatación
Síndrome de Horner: no hay respuesta midriática.
Mecanismo: la cocaína actúa como un agente simpáticomimético por inhibición de la recaptación de norepinefrina en la terminación del nervio. Por lo tanto, la midriasis ocurre en la pupila normal pero no en la pupila de Horner que tiene un déficit de norepinefrina
Observación: para asegurarse la certeza del test los resultados deben evaluarse a los 30 minutos después de administrar cocaína.
Desventaja: sustancia controlada, no hay disponibilidad, y sus metabolitos se detectan en la orina.
Test de la apraclonidina (actual test de elección)
Agente: apraclonidina (0,5%)
Respuesta normal: miosis relativa
Síndrome de Horner: midriasis relativa y recuperación de la ptosis
Mecanismo: la apraclonidina es un débil alfa 1 agonista y un fuerte alfa 2 agonista. En el síndrome de Horner hay una up-regulation de receptores alfa 1 que aumentan la sensibilidad a la apraclonidina. La supersensibilidad de denervación resulta en una dilatación pupilar y elevación del párpado en el lado anormal pero sin respuesta o leve miosis en el lado normal por la actividad alfa 2.
Observación: la apraclonidina está fácilmente disponible con una sensibilidad del 87% (12) Su efecto midriáticoen la pupila normal hace la interpretación mas fácil
Test para localización de la lesión (preganglionar o posganglionar)
Test de la hidroxianfetamina
Agente: hidroxianfetamina (1%)
Respuesta normal: dilatación (lesión preganglionar)
Síndrome de Horner: la no respuesta indica lesión posganglionar
Mecanismo: la hidroxianfetamina promueve la liberación de norepinefrina almacenada de las terminales axónicas posganglionares en la unión neuromuscular del músculo dilatador del iris, significando que si las células posganglionares y sus terminales en el músculo dilatador están intactas, la hidroxianfetamina liberará la norepinefrina almacenada y bloqueará la recaptación, ambas acciones promueven la dilatación.
Observación: al menos 24 hs deben pasar entre la administración de cocaína y el test de hidroxianfetamina debido a que la cocaína tiene la capacidad de inhibir la recaptación de hidroxianfetamina en la vesículas presinápticas que reducirán la certezaTratamiento
Tratamiento medico: hay que tratar la enfermedad de base. Se deben realizar apropiads consultas a especialidades como neumonología, neurología, neurooftalmología, internistas etc) para manejar la enfermedad de base.
Conclusiones: El síndrome de Horner es un complejo signo sintomatológico que se ofrece al clínico como un acertijo, abriendo las probabilidades de entidades causales de lo más variadas. Es así que las causas pueden ir desde un accidente cerebrovascular bulbo-medular lateral (síndrome de Wallenberg), pasando por un traumatismo en el cuello o un cáncer del vértice pulmonar que es el fantasma tan temido, y el peor escenario ante un paciente con este síndrome. Es además, el síndrome de Horner un pretexto para repasar la vía neurológica de la inervación simpática del ojo y de la cara, y un fiel ejemplo de la importancia de la obtención de una correcta historia clínica y un completo examen semiológico.
Por último, cada vez que veamos una ptosis incompleta, como la de nuestra paciente (imagen superior), debemos primero hacer un completo examen semiológico, y eventuales pruebas farmacológicas que nos permitan establecer si se trata o no de un síndrome, de Horner, luego hacer un diagnóstico topográfico de lesión y finalmente un diagnóstico etiológico, para considerar cual es el mejor tratamiento para nuestro paciente
Autor del artículo
Malvinder S Parmar, MB, MS, FRCP(C), FACP, Assistant Professor (VPT), Faculty of Medicine, University of Ottawa; Associate Professor, Department of Internal Medicine, Northern Ontario School of Medicine, Timmins and District Hospital, CanadaMalvinder S Parmar, MB, MS, FRCP(C), FACP is a member of the following medical societies: American College of Physicians, American Society of Nephrology, Canadian Medical Association, Ontario Medical Association, and Royal College of Physicians and Surgeons of Canada Disclosure: Nothing to disclose
Medical Editor
Philip Schulman, MD, Chief, Medical Oncology, Department of Medicine, Memorial Sloan-Kettering Cancer Center; Clinical Professor, Department of Medicine, New York University School of MedicinePhilip Schulman, MD is a member of the following medical societies: American Association for Cancer Research, American College of Physicians, American Society of Clinical Oncology, American Society of Hematology, and Medical Society of the State of New York Disclosure: celgene Honoraria for Speaking and teaching; Amgen Honoraria for Speaking and teaching; genetech/idec Honoraria for Speaking and teaching
Fuente1. du Petit FP. Mémoire dans lequel il est démontré que les nerfs intercostaux fournissent des rameaux que portent des esprits dans les yeux. Paris, Mém: Hist Acad Roy Sci; 1727;1-19.
2. Bernard C. Des phénomènes oculo-pupillaires produits par la section du nerf sympathique cervical: ils sont indépendants des phénomènes vasculaires calorifiques de la tête. Comptes rendus de l'Académie des sciences, Paris. 1852;55:381-88.
3. Weir Mitchell S, Keen Jr W, Morehouse GR. Gunshot Wounds and Other Injuries of Nerves. Philadelphia: Lippincott; 1864. Reprinted, San Francisco: Norman Publishing; 1989.
4. Horner JF. Über eine Form von Ptosis. Klinische Monatsblätter für Augenheilkunde, Stuttgart. 1869;7:193-8.
5. Durham DG. Congenital hereditary Horner's syndrome. AMA Arch Ophthalmol. Nov 1958;60(5):939-40. [Medline].
6. von Passow A. Okulare Paresen im Symptomenbilde des "Status dysraphicus", zugleich ein Beitrag zur Ätiologie der Sympathikusparese (Horner-Syndrom und Heterochromia iridis). Münchener medizinische Wochenshrift. 1934;74:1243-9.
7. Krasnianski M, Georgiadis D, Grehl H, Lindner A. [Correlation of clinical and magnetic resonance imaging findings in patients with brainstem infarction]. Fortschr Neurol Psychiatr. May 2001;69(5):236-41. [Medline].
8. Biousse V, Touboul PJ, D'Anglejan-Chatillon J, Lévy C, Schaison M, Bousser MG. Ophthalmologic manifestations of internal carotid artery dissection. Am J Ophthalmol. Oct 1998;126(4):565-77. [Medline].
9. Raeder JG. Brain. Oxford: 1924:47;149-158.
10. Rombolá CA, Atance PL, Honguero Martínez AF. Claude Bernard-Horner Syndrome as a Rare Complication of Postoperative Pleural Drainage. Arch Bronconeumol. 2008;44:116-7. [Medline].
11. Morales J, Brown SM, Abdul-Rahim AS, Crosson CE. Ocular effects of apraclonidine in Horner syndrome. Arch Ophthalmol. Jul 2000;118(7):951-4. [Medline].
12. Koc F, Kavuncu S, Kansu T, Acaroglu G, Firat E. The sensitivity and specificity of 0.5% apraclonidine in the diagnosis of oculosympathetic paresis. Br J Ophthalmol. Nov 2005;89(11):1442-4. [Medline].
13. Adams RD, Victor M. Eye signs in neurologic diagnosis. In: Principles of Neurology. 5th ed. New York: McGraw-Hill, Inc; 1993:375-9.
14. Biousse V, Touboul PJ, D'Anglejan-Chatillon J, Levy C, Schaison M, et al. Ophthalmologic manifestations of internal carotid artery dissection. Am J Ophthalmol. Oct 1998;126(4):565-77. [Medline].
15. Birch C. Horner. In: Names We Remember 56 Eponymous Medical Biographies. Kent, England: Ravenswood, Beckenham; 70-72.
16. Bucci T, Califano L. Bernard-Horner's syndrome: unusual complication after neck dissection. J Oral Maxillofac Surg. Apr 2008;66(4):833. [Medline].
17. Dziewas R, Konrad C, Drager B, Evers S, Besselmann M, Ludemann P, et al. Cervical artery dissection--clinical features, risk factors, therapy and outcome in 126 patients. J Neurol. Oct 2003;250(10):1179-84. [Medline].
18. Lee JH, Lee HK, Lee DH, Choi CG, Kim SJ, Suh DC. Neuroimaging strategies for three types of Horner syndrome with emphasis on anatomic location. AJR Am J Roentgenol. Jan 2007;188(1):W74-W81. [Medline].
19. Magalini SI, Magalini SC. Dictionary of Medical Syndromes. Philadelphia, Pa: Lippincott Williams & Wilkins; 1997.
20. McCorry D, Bamford J. Painful Horner's syndrome caused by carotid dissection. Postgrad Med J. Mar 2004;80(941):164. [Medline].
21. Merrison AF, Lhatoo SD. Horner's syndrome postpartum. BJOG. Jan 2004;111(1):86-8. [Medline].
22. Morris JG, Lee J, Lim CL. Facial sweating in Horner's syndrome. Brain. Sep 1984;107 ( Pt 3):751-8. [Medline].
23. Nautiyal A, Singh S, DiSalle M, O'Sullivan J. Painful Horner syndrome as a harbinger of silent carotid dissection. PLoS Med. Jan 2005;2(1):e19. [Medline].
24. Piccoli M, Golinelli M, Colli G, Mullineris B, Melotti G. Homer syndrome after thoracoscopic apicectomy for spontaneous pneumothorax as a complication of chest tube placement. Chir Ital. Nov-Dec 2007;59(6):887-9. [Medline].
25. Walton KA, Buono LM. Horner syndrome. Curr Opin Ophthalmol. Dec 2003;14(6):357-63. [Medline].
26. Weiner WJ, Goetz C. Disorders of ocular movement and pupillary function. In: Neurology for the Non-neurologist. 1999. Lippincott, Williams & Wilkins; 4th ed:242-5.
 capular irradiado a región de hombro izquierdo de 3 meses de evolución tratada con medicación antiinflamatoria y corticosteroides sin respuesta.
capular irradiado a región de hombro izquierdo de 3 meses de evolución tratada con medicación antiinflamatoria y corticosteroides sin respuesta.Al ingreso al hospital Pintos, la paciente presenta una anormalidad facial.

Cual es el diagnóstico?
La paciente registraba antecedentes de fumadora importante hasta hace seis años.
En el examen físico se objetivó un síndrome de Horner izquierdo, consistente en ptosis incompleta ,
En el examen físico se objetivó un síndrome de Horner izquierdo, consistente en ptosis incompleta ,

miosis y anhidrosis en la hemicara ipsilateral.
En la fosa supraclavicular izquierda se obsevaba una asimetría comparando con la derecha, y a la palpación se tocaba una zona indurada fija a los planos profundos y polilobulada que impresionaba como adenomegalias.
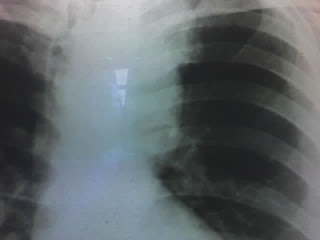
Se realizó una Rx de tórax (imagen) donde se observaba un mediastino alto ensanchado a la izquierda y una TAC que mostró una tumoración en lóbulo superior izquierdo.
En la fosa supraclavicular izquierda se obsevaba una asimetría comparando con la derecha, y a la palpación se tocaba una zona indurada fija a los planos profundos y polilobulada que impresionaba como adenomegalias.
Se realizó una Rx de tórax (imagen) donde se observaba un mediastino alto ensanchado a la izquierda y una TAC que mostró una tumoración en lóbulo superior izquierdo.

Se llevó a cabo una biopsia escisional de adenomegalia de fosa supraclavicular que mostró metástasis de carcinoma indiferenciado de pulmón. En este momento se está realizando inmunomarcación de tejido ganglionar.
Es decir que se trata de un cáncer de pulmón que debutó con dolor dorsal ysíndrome de Horner izquierdo
SINDROME DE HORNER
Antecedentes
Antecedentes

El syndrome de Horner resulta de una interrupción del tono simpático al ojo y se caracteriza por la clásica tríada de miosis, ptosis parcial, y ausencia de sudoración hemifacial (anhidrosis)Origen del términoFrançois Pourfour du Petit (1664-1741) describió primero esta condición en 1727 en animales de experimentación cortando los nervios en el cuello de perros y notando los trastornos que ocurrian en el ojo y la hemicara homolateral a la lesión neurológica. (1) En 1838 el clínico ingles Edgard Selleck Hare (1812-1838) hizo una descripción de un paciente que murió de un tumor en la región cervical. Fue mas profundamente descripto por Claude Bernard en 1852 (2)
Un reporte clínico del síndrome en un hombre que recibió un trauma en el cuello fue descripto en 1864 por los tres clínicos del ejército norteamericano: Silas Weir Mitchell (1829-1914), William Keen Jr. (1837-1932), and George Read Morehouse (1829-1905) (3). Mas tarde, en 1869, Horner describió una mujer de 40 años quien desarrolló las manifestaciones clásicas del síndrome (4) En 1958. D G Dirham documentó una familia en la que 5 personas en 2 generaciones fueron afectados (5) Esta rara condición genética es probablemente un rasgo dominante autonómico.
El término síndrome de Horner es comúnmente usado en paises ingleses, mientras que el término síndrome de Bernard-Horner es común en Francia.
El síndrome de von Passow es una asociación de síndrome de Horner con heterocromía del iris (6)
Patofisiología:La inervación simpática del ojo consiste en una vía de 3 neuronas. La primera neurona está en el hipotálamo ipsilateral y sus axones descienden por el tronco cerebral y la médula cervical hasta T1-T2. Esas fibras hacen sinapsis después de salir de la médula con el ganglio simpático cervical y hasta entonces son llamadas preganglionares. Una vez que hacen sinapsis con las células del ganglio cervical superior, las fibras de tercer orden viajan via carótida interna hasta la órbita, e inervan los músculos dilatadores del iris.
Las fibras simpáticas posganglionares también inervan el músculo de Mueller dentro del párpado. Este músculo es el responsable de iniciar la retracción del párpado durante la apertura palpebral. Las fibras simpáticas posganglionares, responsables de la sudoración facial siguen a la carótida interna hasta las glándulas sudoríparas de la cara. La interrupción en cualquier localización resulta en síndrome de Horner ipsilateral.
El síndrome de Horner puede ser el resultado de las siguientes condiciones:
* Lesión en la primera neurona (hipotálamo).
* Accidente cerebrovascular o tumor de tronco cerebral.
* Trauma de plexo braquial.
* Tumor (por ej Pancoast) o infección del vértice pulmonar.
* Lesión de fibras posganglionares.
* Disección carotídea (en un estudio 44% de los pacientes con disección de carótida extracraneal tuvo síndrome de Horner doloroso).
* Isquemia de la arteria carótida.
* Migraña.
* Neoplasia de fosa craneal media.
Frecuencia:El síndrome de Horner es poco común.
Morbi/mortalidad:Depende de la enfermedad de base.
Raza:No hay prerdilección por ninguna raza
Sexo: no hay predilección por sexo
Edad:No hay predilección por ninguna edad
Clínica:Los síntomas dependen de la enfermedad de base
Los pacientes pueden ser incapaces de abrir completamente el ojo afectado o de presentar trastornos de la sudoración en la hemicara afectada.
Los pacientes conlesiones preganglionares pueden tener enrojecimiento facial (flushing) (efecto arlequin) después de un ejercicio físico.
Los pacientes con lesiones posganglionares pueden tener dolor orbitario ipsilateral o cefalea de tipo “migraña like”
Raeder, un oftalmólogo noruego describió un paciente con combinación de dolor orbitario, miosis, y ptosis, síndrome que denominó paratrigeminal (9)
Cuando se asocia a disección carotídea los pacientes pueden presentarse con dolor ipsilateral en cuello, cabeza y cara
Los signos
Características pupilares:
La pupila del lado afectado puede ser redonda y contraida (miótica)
Los pacientes pueden tener pérdida del reflejo ciliospinal (aferente C2, C3). Las pupilas no se dilatan cuando la piel de la espalda es pinchada. Este hallazgo según muchos autores no es confiable
La anisocoria es mas evidente en la oscuridad. La pupila afectada se dilate mas lentamente que la pupila normal
Características faciales:
Los pacientes tienen la piel seca (anhidrosis) del mismo lado que la pupila afectada
Si la lesión es en la arteria carótida primitiva la pérdida de sudoración afecta toda la cara, y si la lesión es distal a la bifurcación la ausencia de sudoración está restringida a la frente y el lado de la nariz ipsilateral a la lesión
Otros hallazgos:
Los pacientes pueden tener ptosis parcial.
Pueden tener enoftalmos, aunque en general esto no ocurre, y la ptosis da la ilusión de enoftalmos, producido por la debilidad del músculo de Mueller, tanto en el párpado superior (causando ptosis parcial) como en el inferior
La amplitud de la acomodación aumenta.
La heterocromía del iris puede presentarse si la lesión es en un niño menor de 2 años. El iris afectado puede permanecer azul cuando el otro cambia a marrón. La pigmentación del iris está bajo control simpático durante el desarrollo, que es completado a los 2 años. La heterocromía del iris es infrecuente en pacientes con síndrome de Horner adquirido tardiamente en la vida.
Los pacientes pueden tener retracción del párpado contralateral .
Los pacientes pueden presentar disminución transitoria de la presión intraocular y cambios en la viscosidad de las lágrimas.
Los pacientes pueden no tener pliegue horizontal en el párpado ptosado especialmente en aquellos con síndrome de Horner congénito.
Los pacientes pueden tener enrojecimiento de la conjuntiva.
Causas:El síndrome de Horner puede ser congénito, adquirido, hereditario (autonómico dominante) La interrupción de las fibras simpáticas puede ocurrir en sistema nervioso central (entre el hipotálamo y el lugar de emergencia de la médula espinal (C8, T2), o en sistema nervioso periférico (por ejemplo en cadena simpática cervical, ganglio cervical superior, junto a la arteria carótida)
Causas centrales (raras)
El síndrome de Horner puede estar asociado con una lesión en el hipotálamo, hasta la médula en la región cervical superior. El síndrome medular lateral de Wallenberg (stroke), enfermedades desmielinizantes, y mas raramente trauma, o siringomielia pueden resultar en síndrome de Horner.
Es raro en el coma, pero el síndrome de Horner puede ocurrir ipsilateralmente a una gran hemorragia cerebral que afecta el tálamo. El síndrome de Horner ipsilateral en un paciente con parálisis laringea sugiere una lesión intramedular
El síndrome de Horner que ocurre en asociación con trauma medular sugiere lesión cervical alta, y no ocurre en lesiones por debajo de T2 o T3.
Causas periféricas:
El síndrome de Horner puede ser causado por lesiones en la cadena simpática, en el ganglio cervical superior, o acompañando a la arteria carótida como resultado de un tumor de Pancoast (carcinoma del vértice pulmonar), trauma, canulación de la vena yugular interna, o raramente de disección carotidea, trauma carotideo, colocación de un tubo en tórax para drenaje de derrame pleural (10), sarcoidosis, o tuberculosis en los ganglios cervicales (escrófula)
Un síndrome de Horner ipsilateral y neuropatía craneal afectando los pares craneales IX hasta XII puede estar asociado con compromiso carotideo, una rara pero fatal complicación de una infección del espacio faringeo.
La lesión común que causa síndrome de Horner, interfiere con las fibras preganglionares cuando ellas atraviesan la parte superior del tórax. Virtualmente todas las lesiones simpáticas posganglionares están localizadas intracranealmente o infraorbitariamente debido a que el ganglio cervical superior está cerca del cráneo.
El síndrome de Horner preganglionar indica una seria patología y con alta frecuencia se asocia a cáncer. Las causas comunes de síndrome de Horner incluyen trauma, disección aórtica, disección carotidea, y tumor de Pancoast, metástasis a los ganglios cervicales, y sarcoidosis o tuberculosis de los ganglios cervicales.
Un síndrome de Horner doloroso sugiere la posibilidad de disección de carótida interna.
El síndrome de Horner posganglionar tiene causas principalmente benignas (usualmente cefalea vascular). Las causas comunes de síndrome de Horner posganglionar incluyen trauma, cefalea histamínica y cirugía de cuello o de toroides
Diagnóstico diferencial
Anisocoria simple
Miosis senil
Pupila de Argyll Robertson.
Uso de drogas mióticas
Pupila de Adie-Holmes (contralateral)
Métodos de estudioImágenes:
Obtener una Rx de tórax para descartar un tumor del vértice pulmonar (carcinoma broncogénico apical) que es la causa más frecuente de síndrome de Horner.
Realizar TC de cráneo, si se sospecha ACV (stroke).
En el síndrome de Horner doloroso, obtener una angiografía por RMN con imágenes de sección cruzada del cuello para evaluar la posibilidad de disección carotidea
Procedimientos o maniobras diagnósticas
Los siguientes tests farmacológicos documentan la presencia o ausencia de una lesión simpática ocular e identifican el nivel de compromiso (preganglionar o posganglionar). Localizar la topografía de la lesión es importante debido a que las fibras preganglionares están asociadas a una mayor incidencia de cáncer que requiere investigaciones más profundas.
Tests que documentan la lesión ocular simpática
Test de la cocaína tópica:Agente: cocaína (2 a 4%)
Respuesta normal: dilatación
Síndrome de Horner: no hay respuesta midriática.
Mecanismo: la cocaína actúa como un agente simpáticomimético por inhibición de la recaptación de norepinefrina en la terminación del nervio. Por lo tanto, la midriasis ocurre en la pupila normal pero no en la pupila de Horner que tiene un déficit de norepinefrina
Observación: para asegurarse la certeza del test los resultados deben evaluarse a los 30 minutos después de administrar cocaína.
Desventaja: sustancia controlada, no hay disponibilidad, y sus metabolitos se detectan en la orina.
Test de la apraclonidina (actual test de elección)
Agente: apraclonidina (0,5%)
Respuesta normal: miosis relativa
Síndrome de Horner: midriasis relativa y recuperación de la ptosis
Mecanismo: la apraclonidina es un débil alfa 1 agonista y un fuerte alfa 2 agonista. En el síndrome de Horner hay una up-regulation de receptores alfa 1 que aumentan la sensibilidad a la apraclonidina. La supersensibilidad de denervación resulta en una dilatación pupilar y elevación del párpado en el lado anormal pero sin respuesta o leve miosis en el lado normal por la actividad alfa 2.
Observación: la apraclonidina está fácilmente disponible con una sensibilidad del 87% (12) Su efecto midriáticoen la pupila normal hace la interpretación mas fácil
Test para localización de la lesión (preganglionar o posganglionar)
Test de la hidroxianfetamina
Agente: hidroxianfetamina (1%)
Respuesta normal: dilatación (lesión preganglionar)
Síndrome de Horner: la no respuesta indica lesión posganglionar
Mecanismo: la hidroxianfetamina promueve la liberación de norepinefrina almacenada de las terminales axónicas posganglionares en la unión neuromuscular del músculo dilatador del iris, significando que si las células posganglionares y sus terminales en el músculo dilatador están intactas, la hidroxianfetamina liberará la norepinefrina almacenada y bloqueará la recaptación, ambas acciones promueven la dilatación.
Observación: al menos 24 hs deben pasar entre la administración de cocaína y el test de hidroxianfetamina debido a que la cocaína tiene la capacidad de inhibir la recaptación de hidroxianfetamina en la vesículas presinápticas que reducirán la certezaTratamiento
Tratamiento medico: hay que tratar la enfermedad de base. Se deben realizar apropiads consultas a especialidades como neumonología, neurología, neurooftalmología, internistas etc) para manejar la enfermedad de base.
Conclusiones: El síndrome de Horner es un complejo signo sintomatológico que se ofrece al clínico como un acertijo, abriendo las probabilidades de entidades causales de lo más variadas. Es así que las causas pueden ir desde un accidente cerebrovascular bulbo-medular lateral (síndrome de Wallenberg), pasando por un traumatismo en el cuello o un cáncer del vértice pulmonar que es el fantasma tan temido, y el peor escenario ante un paciente con este síndrome. Es además, el síndrome de Horner un pretexto para repasar la vía neurológica de la inervación simpática del ojo y de la cara, y un fiel ejemplo de la importancia de la obtención de una correcta historia clínica y un completo examen semiológico.
Por último, cada vez que veamos una ptosis incompleta, como la de nuestra paciente (imagen superior), debemos primero hacer un completo examen semiológico, y eventuales pruebas farmacológicas que nos permitan establecer si se trata o no de un síndrome, de Horner, luego hacer un diagnóstico topográfico de lesión y finalmente un diagnóstico etiológico, para considerar cual es el mejor tratamiento para nuestro paciente
Autor del artículo
Malvinder S Parmar, MB, MS, FRCP(C), FACP, Assistant Professor (VPT), Faculty of Medicine, University of Ottawa; Associate Professor, Department of Internal Medicine, Northern Ontario School of Medicine, Timmins and District Hospital, CanadaMalvinder S Parmar, MB, MS, FRCP(C), FACP is a member of the following medical societies: American College of Physicians, American Society of Nephrology, Canadian Medical Association, Ontario Medical Association, and Royal College of Physicians and Surgeons of Canada Disclosure: Nothing to disclose
Medical Editor
Philip Schulman, MD, Chief, Medical Oncology, Department of Medicine, Memorial Sloan-Kettering Cancer Center; Clinical Professor, Department of Medicine, New York University School of MedicinePhilip Schulman, MD is a member of the following medical societies: American Association for Cancer Research, American College of Physicians, American Society of Clinical Oncology, American Society of Hematology, and Medical Society of the State of New York Disclosure: celgene Honoraria for Speaking and teaching; Amgen Honoraria for Speaking and teaching; genetech/idec Honoraria for Speaking and teaching
Fuente1. du Petit FP. Mémoire dans lequel il est démontré que les nerfs intercostaux fournissent des rameaux que portent des esprits dans les yeux. Paris, Mém: Hist Acad Roy Sci; 1727;1-19.
2. Bernard C. Des phénomènes oculo-pupillaires produits par la section du nerf sympathique cervical: ils sont indépendants des phénomènes vasculaires calorifiques de la tête. Comptes rendus de l'Académie des sciences, Paris. 1852;55:381-88.
3. Weir Mitchell S, Keen Jr W, Morehouse GR. Gunshot Wounds and Other Injuries of Nerves. Philadelphia: Lippincott; 1864. Reprinted, San Francisco: Norman Publishing; 1989.
4. Horner JF. Über eine Form von Ptosis. Klinische Monatsblätter für Augenheilkunde, Stuttgart. 1869;7:193-8.
5. Durham DG. Congenital hereditary Horner's syndrome. AMA Arch Ophthalmol. Nov 1958;60(5):939-40. [Medline].
6. von Passow A. Okulare Paresen im Symptomenbilde des "Status dysraphicus", zugleich ein Beitrag zur Ätiologie der Sympathikusparese (Horner-Syndrom und Heterochromia iridis). Münchener medizinische Wochenshrift. 1934;74:1243-9.
7. Krasnianski M, Georgiadis D, Grehl H, Lindner A. [Correlation of clinical and magnetic resonance imaging findings in patients with brainstem infarction]. Fortschr Neurol Psychiatr. May 2001;69(5):236-41. [Medline].
8. Biousse V, Touboul PJ, D'Anglejan-Chatillon J, Lévy C, Schaison M, Bousser MG. Ophthalmologic manifestations of internal carotid artery dissection. Am J Ophthalmol. Oct 1998;126(4):565-77. [Medline].
9. Raeder JG. Brain. Oxford: 1924:47;149-158.
10. Rombolá CA, Atance PL, Honguero Martínez AF. Claude Bernard-Horner Syndrome as a Rare Complication of Postoperative Pleural Drainage. Arch Bronconeumol. 2008;44:116-7. [Medline].
11. Morales J, Brown SM, Abdul-Rahim AS, Crosson CE. Ocular effects of apraclonidine in Horner syndrome. Arch Ophthalmol. Jul 2000;118(7):951-4. [Medline].
12. Koc F, Kavuncu S, Kansu T, Acaroglu G, Firat E. The sensitivity and specificity of 0.5% apraclonidine in the diagnosis of oculosympathetic paresis. Br J Ophthalmol. Nov 2005;89(11):1442-4. [Medline].
13. Adams RD, Victor M. Eye signs in neurologic diagnosis. In: Principles of Neurology. 5th ed. New York: McGraw-Hill, Inc; 1993:375-9.
14. Biousse V, Touboul PJ, D'Anglejan-Chatillon J, Levy C, Schaison M, et al. Ophthalmologic manifestations of internal carotid artery dissection. Am J Ophthalmol. Oct 1998;126(4):565-77. [Medline].
15. Birch C. Horner. In: Names We Remember 56 Eponymous Medical Biographies. Kent, England: Ravenswood, Beckenham; 70-72.
16. Bucci T, Califano L. Bernard-Horner's syndrome: unusual complication after neck dissection. J Oral Maxillofac Surg. Apr 2008;66(4):833. [Medline].
17. Dziewas R, Konrad C, Drager B, Evers S, Besselmann M, Ludemann P, et al. Cervical artery dissection--clinical features, risk factors, therapy and outcome in 126 patients. J Neurol. Oct 2003;250(10):1179-84. [Medline].
18. Lee JH, Lee HK, Lee DH, Choi CG, Kim SJ, Suh DC. Neuroimaging strategies for three types of Horner syndrome with emphasis on anatomic location. AJR Am J Roentgenol. Jan 2007;188(1):W74-W81. [Medline].
19. Magalini SI, Magalini SC. Dictionary of Medical Syndromes. Philadelphia, Pa: Lippincott Williams & Wilkins; 1997.
20. McCorry D, Bamford J. Painful Horner's syndrome caused by carotid dissection. Postgrad Med J. Mar 2004;80(941):164. [Medline].
21. Merrison AF, Lhatoo SD. Horner's syndrome postpartum. BJOG. Jan 2004;111(1):86-8. [Medline].
22. Morris JG, Lee J, Lim CL. Facial sweating in Horner's syndrome. Brain. Sep 1984;107 ( Pt 3):751-8. [Medline].
23. Nautiyal A, Singh S, DiSalle M, O'Sullivan J. Painful Horner syndrome as a harbinger of silent carotid dissection. PLoS Med. Jan 2005;2(1):e19. [Medline].
24. Piccoli M, Golinelli M, Colli G, Mullineris B, Melotti G. Homer syndrome after thoracoscopic apicectomy for spontaneous pneumothorax as a complication of chest tube placement. Chir Ital. Nov-Dec 2007;59(6):887-9. [Medline].
25. Walton KA, Buono LM. Horner syndrome. Curr Opin Ophthalmol. Dec 2003;14(6):357-63. [Medline].
26. Weiner WJ, Goetz C. Disorders of ocular movement and pupillary function. In: Neurology for the Non-neurologist. 1999. Lippincott, Williams & Wilkins; 4th ed:242-5.







